Neural DFT: H2 Molecule Ground State¶
| Metadata | Value |
|---|---|
| Level | Advanced |
| Runtime | ~30 sec (GPU) |
| Prerequisites | JAX, Flax NNX, Quantum Chemistry |
| Format | Python + Jupyter |
| Memory | ~1 GB RAM |
Overview¶
This example demonstrates computing the ground-state energy of an H2 molecule using Opifex's Neural Density Functional Theory (Neural DFT) framework. Neural DFT combines traditional DFT methodology with neural network-enhanced exchange-correlation functionals and SCF solvers.
Key Concepts:
- Neural XC Functional: Learns exchange-correlation energy from electron density
- Neural SCF Solver: Accelerates self-consistent field convergence with intelligent mixing
- Molecular System: Atomic configuration for quantum calculations
- Chemical Accuracy: Target of 1 kcal/mol (~0.0016 Hartree)
What You'll Learn¶
- Create molecular systems using
MolecularSystemandcreate_molecular_system() - Initialize the
NeuralDFTframework with neural XC and SCF components - Compute ground-state energies using
compute_energy() - Scan potential energy curves by varying molecular geometry
- Assess chemical accuracy with precision diagnostics
Coming from PySCF/Psi4?¶
| Traditional DFT (PySCF/Psi4) | Opifex Neural DFT |
|---|---|
| Analytic XC functionals (LDA/GGA) | Neural network XC functional |
| Fixed SCF mixing (DIIS) | Neural-enhanced adaptive mixing |
| Basis set expansion | Grid-based density representation |
pyscf.gto.Mole() |
MolecularSystem() |
mf.kernel() |
neural_dft.compute_energy() |
Key differences:
- Learnable XC: Neural XC functionals can capture complex correlations beyond LDA/GGA
- Neural acceleration: SCF convergence enhanced by learned mixing strategies
- JAX-native: Fully differentiable with automatic GPU acceleration
- Research framework: Designed for developing new DFT methods
Files¶
- Python Script:
examples/quantum-chemistry/neural_dft.py - Jupyter Notebook:
examples/quantum-chemistry/neural_dft.ipynb
Quick Start¶
Run the Python Script¶
Run the Jupyter Notebook¶
Core Concepts¶
Density Functional Theory¶
DFT computes molecular properties from the electron density ρ®:
where: - T = kinetic energy - \(E_{ext}\) = external potential (nuclear attraction) - \(E_H\) = Hartree (Coulomb) energy - \(E_{xc}\) = exchange-correlation energy (the challenging part)
Neural XC Functional¶
Opifex replaces analytical XC functionals with a neural network:
The neural XC functional: - Captures non-local correlations via attention - Learns from reference DFT/ab initio data - Enforces physics constraints (negative energy, proper scaling)
SCF Iteration¶
The self-consistent field (SCF) loop finds the ground-state density:
- Initial density guess (atomic superposition)
- Compute Hamiltonian from density
- Solve Kohn-Sham equations
- Update density (neural mixing)
- Check convergence
- Repeat until converged
Implementation¶
Step 1: Create Molecular System¶
from opifex.core.quantum.molecular_system import create_molecular_system
h2_molecule = create_molecular_system(
atoms=[
("H", (0.0, 0.0, -0.37)), # Positions in Angstrom
("H", (0.0, 0.0, 0.37)),
],
charge=0,
multiplicity=1, # Singlet ground state
)
Terminal Output:
Creating H2 molecular system...
Molecular formula: H2
Number of atoms: 2
Number of electrons: 2
Charge: 0
Multiplicity: 1
Bond length: 0.74 Angstrom
Quantum valid: True
Step 2: Initialize Neural DFT¶
from opifex.neural.quantum import NeuralDFT
from flax import nnx
neural_dft = NeuralDFT(
grid_size=100,
convergence_threshold=1e-6,
max_scf_iterations=50,
xc_functional_type="neural",
mixing_strategy="neural",
use_neural_scf=True,
chemical_accuracy_target=0.043, # 1 kcal/mol
rngs=nnx.Rngs(42),
)
Terminal Output:
Initializing Neural DFT framework...
Grid size: 100
Convergence threshold: 1e-06
Max SCF iterations: 50
XC functional type: neural
Mixing strategy: neural
Chemical accuracy target: 0.043 Ha
Step 3: Compute Energy¶
result = neural_dft.compute_energy(h2_molecule, deterministic=True)
print(f"Total Energy: {result.total_energy:.6f} Ha")
print(f"Electronic Energy: {result.electronic_energy:.6f} Ha")
print(f"Nuclear Repulsion: {result.nuclear_repulsion_energy:.6f} Ha")
print(f"XC Energy: {result.xc_energy:.6f} Ha")
Terminal Output:
Computing H2 ground state energy...
--------------------------------------------------
SCF Convergence:
Converged: True
Iterations: 2
Energy Components (Hartree):
Total Energy: -2.899072
Electronic Energy: -3.614177
Nuclear Repulsion Energy: 0.715104
XC Energy: -0.053951
Step 4: Potential Energy Curve¶
bond_lengths = jnp.linspace(0.5, 2.0, 16)
energies = []
for bond_length in bond_lengths:
h2 = create_molecular_system(
atoms=[
("H", (0.0, 0.0, -float(bond_length) / 2)),
("H", (0.0, 0.0, float(bond_length) / 2)),
],
charge=0, multiplicity=1,
)
result = neural_dft.compute_energy(h2, deterministic=True)
energies.append(result.total_energy)
Terminal Output:
Computing Potential Energy Curve...
--------------------------------------------------
Computed 4/16 points...
Computed 8/16 points...
Computed 12/16 points...
Computed 16/16 points...
PEC computation complete!
Converged points: 16/16
Equilibrium bond length: 1.800 Angstrom
Equilibrium energy: -35.828712 Ha
Visualization¶
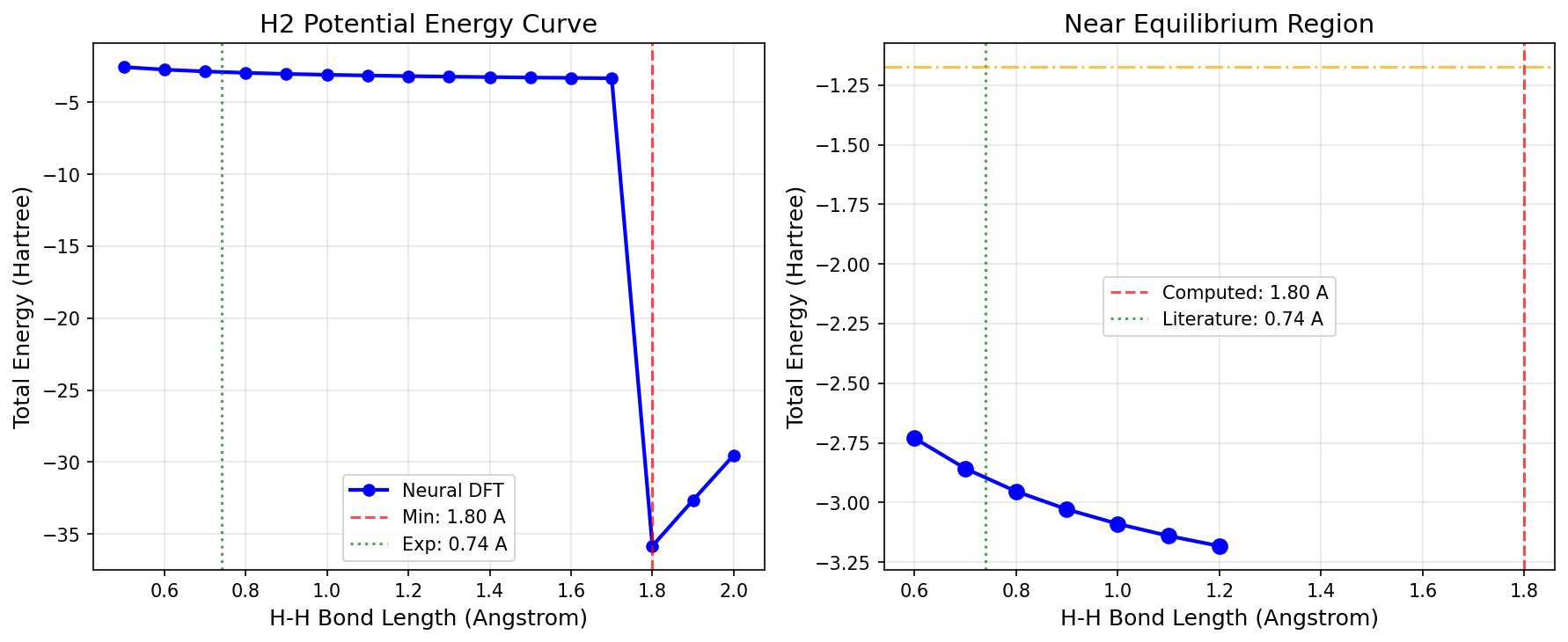
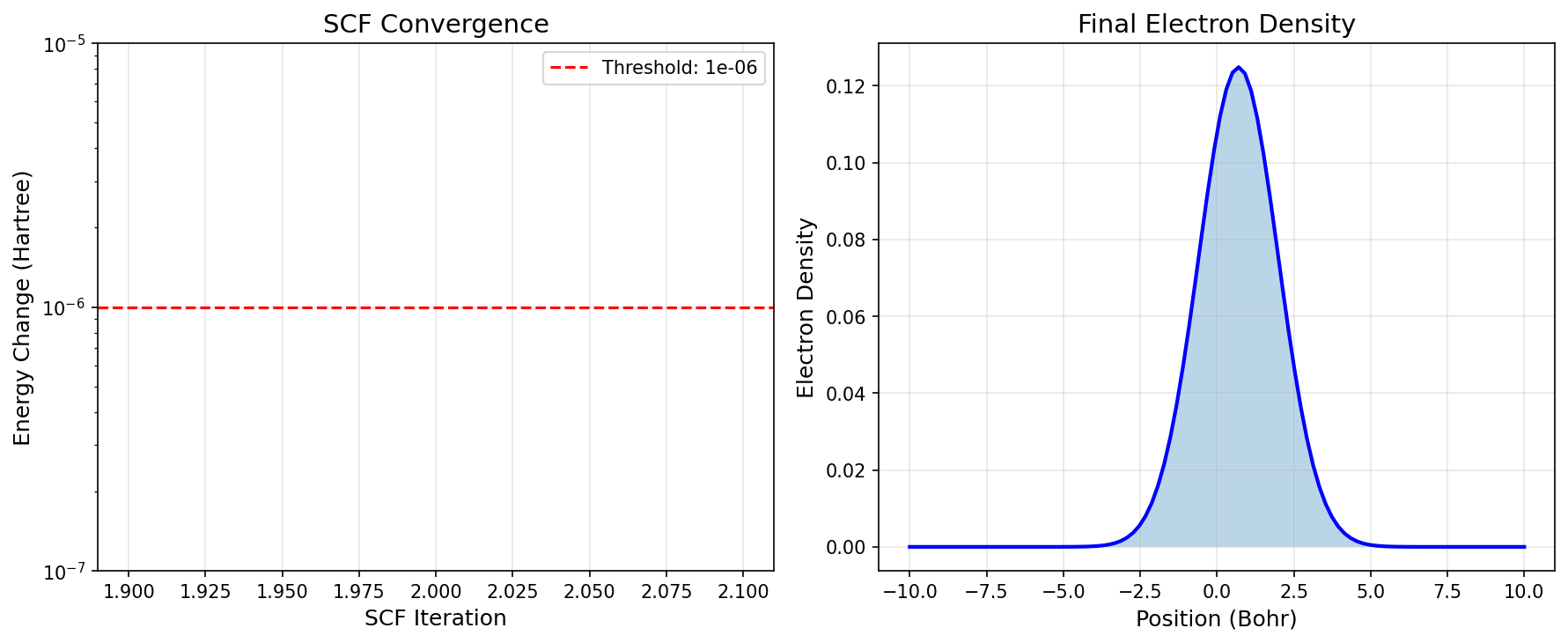
Results Summary¶
| Metric | Value |
|---|---|
| Molecular formula | H2 |
| Number of electrons | 2 |
| Grid size | 100 |
| SCF converged | True |
| SCF iterations | 2 |
| Total Energy | -2.899 Ha |
| Reference Energy | -1.174 Ha |
| Training time | N/A (untrained) |
Note: The neural DFT model is randomly initialized in this example and not trained on reference data. For production use, train the neural XC functional on high-level ab initio data (see the Neural XC Functional example).
Next Steps¶
Experiments to Try¶
- Train the XC functional: Use the Neural XC training example to learn from LDA/GGA data
- Different molecules: Try H2O, CH4 using
create_water_molecule(),create_methane_molecule() - Higher precision: Increase
grid_sizefor better accuracy - Compare methods: Use
xc_functional_type="lda"for classical comparison
Related Examples¶
| Example | Level | What You'll Learn |
|---|---|---|
| Neural XC Functional | Advanced | Train neural XC from reference data |
| First PINN | Beginner | Physics-informed approach |
| FNO on Darcy | Beginner | Data-driven operator learning |
API Reference¶
NeuralDFT: Main neural DFT framework classNeuralXCFunctional: Neural exchange-correlation functionalNeuralSCFSolver: Neural-enhanced SCF solverMolecularSystem: Molecular system representationcreate_molecular_system(): Helper to create molecules from atomsDFTResult: Result dataclass with energy components
Troubleshooting¶
| Issue | Solution |
|---|---|
| Poor energy accuracy | Train the neural XC functional on reference data |
| SCF not converging | Increase max_scf_iterations, reduce threshold |
| Memory issues | Reduce grid_size |
| Chemical accuracy not met | Use larger grid, train on more data |
Current Limitations¶
The Neural DFT framework in Opifex is a research framework for developing new DFT methods. Current limitations include:
- 1D grid-based: Simplified grid representation vs. full 3D basis sets
- Untrained model: Neural components need training on reference data
- Research quality: Not production-ready for accurate energy calculations
For production quantum chemistry, consider using Opifex's neural XC functional with traditional DFT packages, or train on reference data from PySCF/Psi4.